


Have a question about KIF1A-Associated Neurological Disorder? Find answers to Frequently Asked Questions (FAQs) here.
Translate this page:
Are you new to the KIF1A community? Do you have lingering questions about KIF1A? You’ve come to the right place! We will update this page frequently, so make sure you check back often. If you have more questions, send them to us at impact@kif1a.org!
This resource is for informational purposes only and should not be construed as medical advice or used to replace guidance from your healthcare providers. KAND does not affect any two people the same way, so it is important to discuss your specific questions and concerns with healthcare providers who are familiar with your medical history and status.
Last updated February 25, 2021
Most Frequently Asked Questions
KIF1A-Associated Neurological Disorder (KAND) is a rare neurodegenerative disorder caused by one or more changes in the KIF1A gene. When KIF1A.ORG was founded in 2017, this disorder was so rare that it didn’t have a name! Before KAND was named, researchers used different terms to describe people affected by mutations in the KIF1A gene, such as KIF1A-related disorder. You may see the terms KIF1A and KAND used interchangeably in informal contexts.
Genes provide instructions for creating proteins that play a critical role in many functions of the body. KIF1A is the gene responsible for production of the KIF1A protein. The KIF1A protein is part of the kinesin superfamily proteins (KIFs), a type of molecular motor proteins located within the nervous system that are responsible for transporting important cargo, such as nutrients and other molecules, within nerve cells along cellular roadways called microtubules. Motor proteins use a cellular fuel source, adenosine triphosphate (ATP), to transport cargo inside of cells. The KIF1A protein has been called the “molecular marathon runner” because it has to move cargo a very long distance at a very fast speed. A more detailed description of what the KIF1A protein is, how it transports cargo through our nerve cells and how mutation of a KIF1A gene can influence the function of the KIF1A protein can be accessed here:
Depending on the significance of impact the gene variance has on the function of the KIF1A protein, it may be identified as pathogenic, or disease causing. KIF1A-Associated Neurological Disorder, or KAND, is a rare, neurodegenerative disorder caused by pathogenic KIF1A variants. To better understand KAND, check out this short, easy-to-understand video featured below:
KIF1A-Associated Neurological Disorder, or KAND, is a rare, neurodegenerative disorder caused by pathogenic (disease-causing) variants in the KIF1A gene. KAND can only be diagnosed through genetic testing as many symptoms are often mistaken for cerebral palsy or other more common diseases.
Signs and symptoms for KAND vary dramatically from person to person; however, there are common associated symptoms for those living with KAND. Some common symptoms are listed below, but it does not mean an affected individual will experience all of these symptoms.
- Movement
- Hypotonia
- Hypertonia
- Peripheral neuropathy
- Hereditary Spastic Paraplegia (HSP)
- Ataxia
- Seizures/Epilepsy
- Absence
- Tonic/Clonic (Grand mal)
- Atonic
- Cognitive/Behavioral
- Developmental delay/intellectual disability
- Autism
- ADHD
- Anxiety
- Gastrointestinal
- GERD
- Constipation and/or diarrhea
- Ophthalmic (Eyes/Vision)
- Optic nerve atrophy/hypoplasia
- Cortical visual impairment
- Strabismus
- Nystagmus
- Autonomic
- Difficulty with temperature regulation (e.g. sporadic fevers unrelated to illness)
- Decreased pain sensitivity
- Neuroimaging
- Abnormal MRI finding
- Cerebellar and/or cerebral atrophy
Families and clinicians can find additional clinical information on KAND based on our Natural History Study with 177 patients in 2024 paper by Sudnawa et al. Additionally you can read the initial Natural History Study, a 2021 publication from Boyle, et al. with 100 participants.
What is KAND? is a helpful resource explaining the basics of KAND and the common symptoms associated with the disease.
You are not alone. Please visit our Newly Diagnosed page for helpful information and support.
Receiving this diagnosis can have a significant impact, not only on the person receiving the diagnosis, but on others who love and support them. KIF1A.ORG has put together a list of resources available to families as they process this diagnosis. We welcome you to explore resources available to you and your family. Please visit our page for newly diagnosed families. Our Family Support & Resources page offers resources for school, coping with the diagnosis, and understanding KAND for parents and siblings. If you’re looking for something specific but can’t find it here, reach out to us at impact@kif1a.org.
KAND is a neurodegenerative disorder with a progressive course, meaning the condition advances over time. However, the symptoms experienced, severity of symptoms, and progression will vary by individual. Some individuals may not outwardly notice any regression, while others see noticeable changes, such as loss in vision or declining motor skills. For example, in childhood an individual may require assistive devices to walk (such as a walker or crutches), but require a wheelchair by their 20s. Other individuals may experience severe symptoms early in life that prevents them from reaching milestones such as sitting up unassisted or crawling. In other words, while KAND can cause individuals to lose skills or abilities over time, KAND can prevent individuals from developing certain skills or abilities at all, and can lead to early death. The progressive course of KAND underscores our urgency to discover treatments and ultimately a cure.
Although different variations of KIF1A can lead to many of the same signs and symptoms from one person to another, the specific symptoms that develop, the progression of the symptoms and the overall severity varies greatly. As Dr. Wendy Chung explains, “KAND can best be thought of as a spectrum of disease that can range from mild symptoms to severe, life-threatening complications.”
KAND is still not fully understood by the scientific community. In the latest publication (Boyle et al., January 2021) on the KIF1A Natural History Study, patients ranged in age from 6 months to 39 years old. As more adults are identified living with KAND and researchers are able to follow affected individuals over time through the KIF1A Natural History Study, we hope to better answer this question. The answer may also expand over time as standards of care continue to evolve and as treatments hopefully become available. Dr. Chung’s interview about the progressive nature of this disease with Check Rare can be viewed below.
KAND can cause life-threatening complications, such as seizures and respiratory failure due to illness such as pneumonia or influenza. As each patient is unique, it’s important to talk to your doctors about your/your child’s symptoms and risks.
Genetic reports are often filled with highly scientific jargon, making it very difficult to understand without a more simplified explanation. We can help! Though genetic counselors may be available to your family to discuss your genetic report, the image below provides a brief description of information typically found on a genetic report. The format and content available in a genetic report will vary, so yours may not look exactly like the example below.
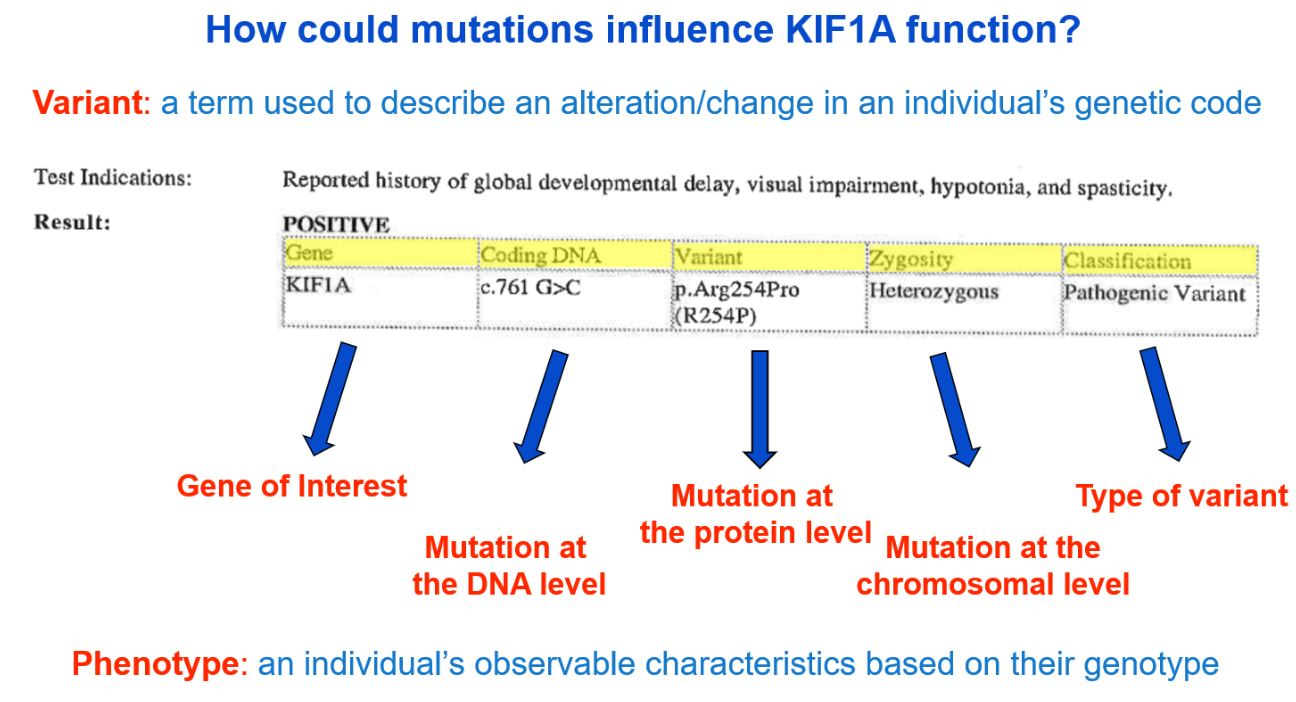
A more detailed description of the image above can be viewed at minute 9:06 here: What Is KIF1A? A Basic Science Explanation for KIF1A Families – YouTube. If you need additional help understanding your genetic report, please email us at impact@kif1a.org!
Variant Classification
A genetic report will classify the variant identified. As you see in the sample genetic report above, the “Classification” indicates that the variant is of a pathogenic (disease-causing) classification. Other possible classifications are “likely pathogenic,” “uncertain significance,” “likely benign” or “benign.”
Variant of Uncertain or Unknown Significance Classification
If you receive a genetic report identifying a KIF1A gene variance but it is classified as a variant of “Unknown Significance” or “Uncertain Significance” (also called “VUS”) please contact Chung Lab by email at kif1a_study@cumc.columbia.edu to discuss the findings further to help evaluate whether the KIF1A variant could be contributing to the symptoms of the patient.
While many variants can cause KIF1A-Associated Neurological Disorder (KAND), variants of “Unknown/Uncertain Significance” may not be the best explanation for a patient’s symptoms. Not all gene variants cause disease. Some patients with a KIF1A variant of unknown/uncertain significance may require further testing and/or evaluation to continue their search for the actual underlying cause of their symptoms. Ask your genetic counselor or physician for guidance on your next steps. Despite any uncertainty on your genetic report, the KIF1A.ORG community is here to support you and help however we can.
Genes, which are made of DNA, code for proteins. In our case, the KIF1A gene gives our bodies instructions to make the KIF1A protein. Changes in DNA can result in changes to how the KIF1A protein is constructed. Variants can be described as changes at the DNA level or protein level using a specific naming system that—at first glance—may look like a bunch of random letters and numbers. Your genetic report will likely include a sequence starting with a “c.” and a sequence starting with a “p.”
- The “c.” sequence describes the “coding DNA reference sequence,” or where the change occurred on the DNA level.
- The “p.” refers to the change at the protein level.
From the genetic report example below, the c. sequence is c.761 G>C, meaning at the 761th position on the KIF1A gene, the nucleotide changed from a G to a C. The variant reported at the p. level is “p.Arg254Pro,” meaning at the 254th amino acid position of the KIF1A gene, the amino acid changed from arginine (abbreviated as “Arg” or “R”) to proline (abbreviated as “Pro” or “P”).
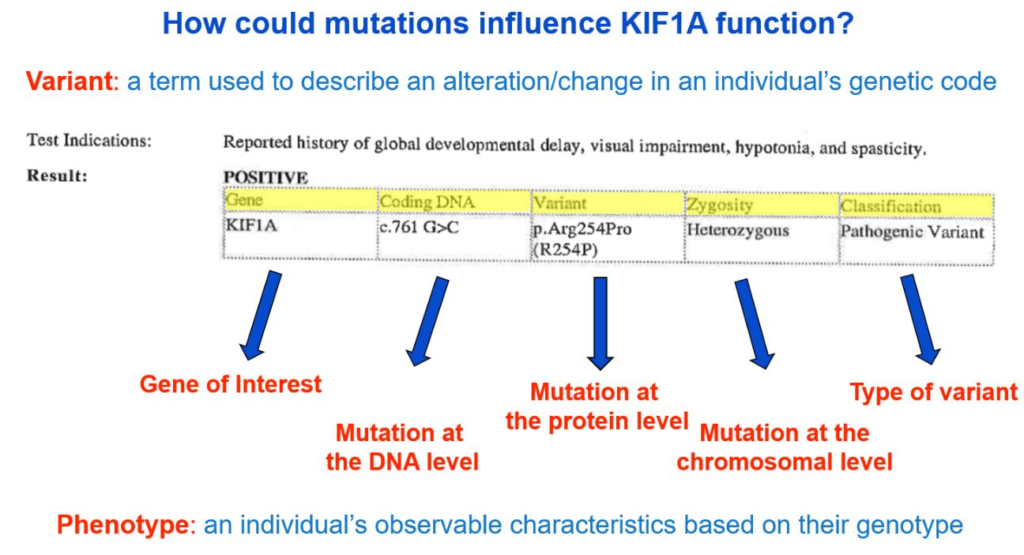
Describing the change at both the coding DNA level and protein level is very useful, depending on the question that is being asked. However, we tend to default to the protein level information when discussing and understanding mutations—so why is that?
While the change at the coding DNA level tells us about the mutation in our genetic code, it doesn’t give us as much insight as to how this change will influence KIF1A function at the protein level. While the DNA level is very important from a genetic standpoint, we are ultimately most interested in how the KIF1A protein is changing as a result of this mutation.
When we look at this change at the protein level, we are talking about amino acids. Amino acids are the building blocks of our proteins—like the individual bricks that, when stacked together, build a greater structure (the KIF1A protein). Amino acids are also made from DNA (DNA is the first step in the process for making an amino acid) which is why a mutation at the DNA level translates to a mutation at the amino acid level.
By talking about mutations at the protein level, we gain important insight as to how the “brick work,” or amino acid, has changed as a result of a mutation in the DNA. This is a concept that is commonly referred to as “structure function”: how does the specific structure of a protein alter its function? Or in the case of KIF1A mutations, how does changing the structure of the KIF1A protein (because of an amino acid change) change the way in which KIF1A works in our bodies?
Note that the example used above (c.761 G>C | p.Arg254Pro) is a missense mutation (a single nucleotide change results in an amino acid change). While the majority of known KAND mutations are missense variants, other changes such as duplications and deletions are possible.
While there is no treatment or cure for KAND yet, the main goal for treating KAND symptoms is “care until a cure.” Below is an excerpt from information provided by Dr. Wendy Chung and Lia Boyle, who lead KAND clinical research, to the NORD Rare Disease Database:
“The treatment of KIF1A-related disorder is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, physicians who specialize in the diagnosis and treatment of neurological disorders in children (pediatric neurologists), neurologists, physicians who specialize in the diagnosis and treatment of eye disorders (ophthalmologists), speech pathologists, physical therapists, and other healthcare professionals may need to systematically and comprehensively plan treatment. Genetic counseling is recommended for affected individuals and their families. Psychosocial support for the entire family is essential as well.
There are no standardized treatment protocols or guidelines for affected individuals. Due to the rarity of the disease, there are no treatment trials that have been tested on a large group of patients. Various treatments have been reported in the medical literature as part of single case reports or small series of patients. Treatment trials will be very helpful to determine the long-term safety and effectiveness of specific medications and treatments for individuals with KIF1A-related disorder.
Following an initial diagnosis, a developmental assessment may be performed and appropriate occupational, physical, speech and feeding therapies be instituted. Periodic reassessments and adjustment of services should be provided with all children. Additional medical, social, and/or vocational services including specialized learning programs may be necessary.”
In addition to a supportive care team and an individualized plan of care, some KAND patients benefit from medications that help treat specific symptoms. Medications, such as gabapentin, may be prescribed to help treat peripheral neuropathy. Baclofen is a muscle relaxer that can help with muscle pain and spasticity. Additionally, Dysport or Botox injections may be used to treat lower-limb spasticity in children. Anti-seizure medication and other medications or treatments targeting specific symptoms may also be prescribed when necessary.
Orthopedic devices including orthotics can aide in stabilizing and improving movement of parts of the body such as knees, ankles, feet, and back. Assistive devices like crutches, canes, and wheelchairs may assist in walking and proper support for mobility. Services to provide support for autism spectrum and/or attention deficit disorders may help those with KAND function in school, work and other areas of life.
Many children with KAND may also benefit from hippotherapy (horseback riding therapy), aquatic therapy, and other evidence-based practices and rehabilitative services. These type of services promote functional outcomes of a task such as increased ability to complete activities of daily living or mobility through increased range of motion or muscle strengthening.
Once a formal diagnosis of a KIF1A gene mutation is confirmed, medical professionals may suggest additional tests to assess the extent of the disease, or if and how the patient is affected by KIF1A-Associated Neurological Disorder (KAND). Some recommendations found in literature and recommendations based on symptoms presented are listed below. As always, please consult with the patient’s healthcare providers to develop a care plan. While there is no treatment or cure for KAND yet, the main goal with treating KAND symptoms is “care until a cure.”
An eye exam with an ophthalmologist is recommended to check for impacts from the disease, such as optic nerve atrophy, cortical visual impairment, strabismus, and cataracts. Individuals may be referred to see an eye doctor, called a neuro-ophthalmologist, specializing in neurological conditions that affect the eyes. Due to the progressive nature of KAND, vision changes can develop over time. Patients may need to schedule regular appointments (e.g. annually or every six months) to monitor for possible changes.
Regular neurological examinations to identify any new symptoms and monitor current issues is vital for people living with KAND. During these routine appointments, laboratory tests or imaging may be ordered.
It is recommended that a baseline 24-hour electroencephalogram (EEG) be completed. An EEG records the brain’s electrical activity and can detect seizures that might not be obvious, as well as detect abnormal brain activity that is not a seizure. It should also be considered as an annual test for neurological monitoring. Since seizures and abnormal brain activity during sleep are common, longer electroencephalograms (EEG) that last 24-48 hours should be considered.
Since atrophy of the brain can be caused by KAND, it may be recommended that a baseline magnetic resonance imaging (MRI) be completed and then routinely scheduled as recommended by your healthcare provider for monitoring of any changes.
Depending on symptoms exhibited, more specific assessments, exams or procedures may be ordered. Evaluations and assessments for rehabilitative services, medical devices, supportive services, special medicine (e.g. orthotist, orthopedist), etc., may be requested/ordered based on needs identified by the KAND patient’s care team.
The simple answer to this question is: we don’t know. We do know that KAND is drastically misdiagnosed and underdiagnosed. Though KIF1A.ORG knows of roughly 550+ individuals throughout the world documented with KIF1A gene mutations, it is likely that the incidence of this disease is in the tens of thousands. Many have either been misdiagnosed with disorders exhibiting similar symptoms or have not yet received a diagnosis. In fact, approximately 1 in 4 of those diagnosed with KIF1A mutations previously had a cerebral palsy diagnosis. Other common misdiagnoses include Rett syndrome and Charcot-Marie-Tooth disease. KIF1A gene mutations are difficult to diagnose without extensive genetic testing, and even with a confirmed diagnosis through testing, families may not be connected with KIF1A.ORG.
Thankfully, there has been exponential growth in the patient population due to advancement of genetic testing, increased access, and relentless advocacy done in collaboration with KIF1A families and research partners. More specialists are now able to learn more and share information about KIF1A.ORG with newly diagnosed families. Though genetic testing has advanced over the years and more individuals are being diagnosed with KAND, genetic testing is still very costly and not accessible to every family that needs it.
If your family has received a KIF1A diagnosis, you can join a global Patient Registry and Natural History Study managed by Chung Laboratory at Boston Children’s Hospital. Your participation is vital to better understanding the disorder and finding treatments. Learn more on our Natural History Study page.
Understanding KAND and the progression of the disease advances the scientific community and our efforts to urgently discover treatment. An essential part of understanding this disease is following KAND patients over time. With funding from KIF1A.ORG, Chung Lab at launched the KIF1A Natural History Study in 2017. It is essential for every KAND family to participate in this voluntary study so researchers can better understand the function of KIF1A. It also allows them to systematically collect and analyze data to inform our path to better care and treatment. Learn more about the Natural History Study and how you can participate.
Financial support is vital to accelerating this urgent research. KIF1A.ORG exists to accelerate discovery of treatments for this generation of people affected by KAND. In addition to ongoing R&D collaborations, KIF1A.ORG funds strategic projects powered to rapidly develop treatment for KAND. The global KIF1A community is highly active in advocacy and raising funds for research. There are many ways to get involved and support our mission.
Please visit our page for newly diagnosed families to learn about other ways to engage with our community.
There has been an uptick in research studies and literature about KIF1A and KAND. Recent and past studies specific to KIF1A and KAND research can be accessed here.
In an effort to make important literature accessible to families, KIF1A.ORG has collaborated with scientists to create “simplified” summaries of their research and scientific concepts that impact our community. You can access Research Simplified summaries here.
The most comprehensive study of KAND to date was led by our very own Lia Boyle and Dr. Chung at the Chung Lab. This research was not only funded by KIF1A.ORG, but it also exists because of the participation from our family community in the Natural History Study. This study is now published in a peer-reviewed journal, which allows physicians and researchers to access and gain a better understanding of KAND. A “Research Simplified” summary of the research paper will be available in the near future. The paper can be accessed here.
The KIF1A Glossary is intended to help KIF1A families understand scientific and clinical terms encountered in research, literature, genetic reports and other scientific communication.